In een informatieve en indrukwekkende documentaire 'De Volendammer ziekte' van de KRO-NCRV [1] worden gezinnen met kinderen met de ernstige erfelijke ziekte PCH2 gefilmd.
 |
| patient met de Volendammer ziekte PCH2 |
Kinderen met PCH2 (wetenschappelijke benaming voor de Volendamse ziekte) hebben slecht ontwikkelende hersenen, waardooor het hoofd klein blijft, hebben een ernstige verstandelijke beperking, meestal leren
ze nooit zitten, staan, lopen, praten en overlijden meestal voor het 10e levensjaar. Ze hebben constant verzorging nodig. De programmamakers hebben ouders gevonden die openhartig wilden vertellen over hun gehandicapt kind en wat het met hen doet. Ik kan de documentaire aan iedereen aanbevelen die wil zien hoe het in de praktijk van een aantal Volendamse gezinnen met zo'n ernstig gehandicapt kind toegaat en wat de houding van die ouders is ten opzichte van DNA diagnostiek. De ouders zouden een eventuele volgende zwangerschap prenataal onderzoek willen laten doen.
Ik wil het in dit blog hebben over de genetische achtergrond van deze ziekte. Volgens de wikipedia [3] is het een autosomaal recessieve aandoening die keurig volgens de wetten van Mendel overerft. Niets bijzonders tot zover. Maar toen ik me ging verdiepen in de genetische achtergrond viel ik van de ene verbazing in de andere. Ik was verbijsterd over de aard en de impact van deze mutatie. Zoiets was ik nog nooit eerder tegengekomen.
Een standaard mutatie zoals wij die kennen is de puntmutatie: de vervanging van één base in het DNA door een andere, met als gevolg dat 1 aminozuur in een eiwit wordt vervangen door een ander aminozuur. Het is gebleken dat de meeste Volendammer patiëntjes homozygoot zijn voor een puntmutatie in positie 307 van het enzym TSEN54 waarbij het amionozuur Alanine vervangen is door Serine. Nu wordt het interessant: TSEN54 is een 'tRNA splicing endonuclease'. Splicing is het verwijderen van een intron. (tRNA is transferRNA). Totaal nieuw en onverwacht voor mij is dat vele tRNA genen óók introns bevatten. tRNAs belichamen in feite de genetische code. Ze zijn de 'vertaalsleutels'. Ze 'vertalen' bijvoorbeeld het triplet AAA in het DNA naar het aminozuur Lysine in het eiwit, enzovoort. Zonder deze 'vertaling' kun je geen eiwit produceren. En eiwitten zijn onmisbaar voor het leven.
Als je ergens géén mutatie in wilt hebben dan is het wel in je tRNA genen. De noodzaak van het verwijderen van een intron vormt een risico. Het kan fout gaan. En in dit geval is de impact groot. Een mutatie in een enzym dat er voor zorgt dat er een normaal tRNA geproduceerd wordt heeft gevolgen voor de productie van alle eiwitten waarin de betreffende aminozuren in voorkomen. Zo heeft de Volendammer mutatie in TSEN54 fatale gevolgen voor de synthese van eiwitten die de volgende 7 aminozuren bevatten: Proline, Arginine, Leucine, Isoleucine, Tyrosine, Cysteine, Tryptofaan. Als een eiwit veel van deze aminozuren bevat moet de impact dramatisch zijn. Als je die inkleurt in de bekende genetische code tabel dan krijg je:
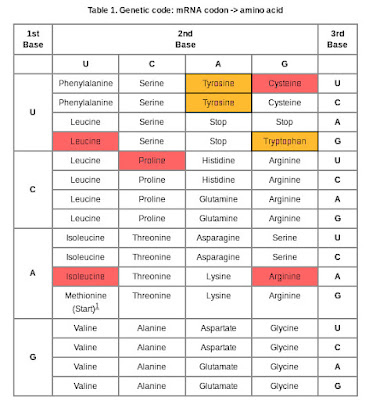 |
Genetische code tabel met 7 Volendammer mutaties:
rood: 5 aminozuren met alternatieve codons
oranje: Tyrosine en Tryptophan zonder alternatieve coderingen |
Wat opvalt is dat 5 aminozuren Leucine, Isoleucine, Proline, Cysteine, Arginine, alternatieve codons hebben. Maar beide codons voor Tyrosine zijn aangetast, en van Tryptofaan is het enige codon aangetast. In dit geval helpt de redundantie in de genetische code helemaal niets. Want die 7 codons in het DNA kunnen überhaupt niet vertaald worden. Alsof een deel van je DNA niet gelezen kan worden. Einde verhaal. Redundantie of niet. Als die codons op belangrijke plaatsen voorkomen in het eiwit, of in het begin, zal dat fataal zijn voor de productie van een compleet eiwit. Een standaard puntmutatie is minder erg dan deze, want de standaard puntmutatie wordt tenminste nog vertaald naar een gewijzigd eiwit en is beperkt tot maar één specifiek eiwit. Bij de Volendammer mutatie zijn potentieel alle eiwitten betrokken die één of meerdere van die 7 aminozuren bevatten, en dat kunnen er honderden of duizenden zijn. Ik zeg 'potentieel', want er is meer aan de hand:
 |
Alle tRNAs en hun aminozuren betrokken bij Volendammer mutatie.
horizontale as: aantal genen; rood: alle tRNAs met introns;
groen: resterende tRNAs zonder introns.
Cysteine (Cys) en Isoleucine (Ile) hebben géén intron-vrij tRNAs.
(eigen grafiek gebaseerd op data uit: [2] )
Dus: 5 van de 8 codons hebben tRNA-gen duplicaten, 3 van de 8 niet.
(update met dank aan Marleen) |
Er is nl. nog een tweede soort redundantie in de genetische code waarvan ik me niet bewust was voordat ik me verdiepte in de Volendammer ziekte. Er zijn meerdere kopieën van tRNA genen in het menselijk genoom ('reserve' of backup kopieën). De kopieën verschillen onderling enigszins, maar hebben dezelfde functie. Een geluk bij een ongeluk is dat er ook tRNA genen zijn zonder introns (groen in de grafiek). Die groene zijn de redding, voor zover ze aanwezig zijn. Omdat daar geen introns verwijderd hoeven worden, worden ze niet beïnvloed door de TSEN54 mutatie. Die normale tRNAs (groene) kunnen de tRNAs die wel getroffen zijn (de rode) compenseren. Ze hebben dezelfde functie. Ze kunnen hun functie overnemen. We zien in de grafiek dat bijvoorbeeld Tryptofaan (Trp) door nog 8 intronloze en dus functionele tRNA genen gecodeerd wordt.
Maar, zoals ook blijkt uit de grafiek worden Cysteïne (Cys) en Isoleucine (Ile) in het geheel niet, en Tyrosine (Tyr) slechts gedeeltelijk gecompenseerd. De TSEN54 mutatie treft twee codons voor Tyrosine (Tyr). De ene heeft één alternatief functioneel tRNA gen, het tweede codon niet. Dat betekent dat het tweede codon voor Tyrosine (Tyr) in het geheel niet vertaald kan worden. Ik kan me niet voorstellen dat het erger dan dat kan zijn.
Er is nog iets positiefs te vertellen. Er zijn maar 32 tRNA genen die het TSEN54 splicing systeem nodig hebben. De mens heeft in totaal 506 tRNA genen. Die genen gebruiken een ander splicing systeem. Dus: de schade die TSEN54 aan kan richten is beperkt tot die 32 genen. Maar desondanks kan de schade aanzienlijk zijn. Dat komt -denk ik- vooral door de drie codons die niet gecompenseerd worden.
Dus: de standaard redundantie in de genetische codering helpt ons hier niet, en er is geen voldoende redundantie in de vorm van tRNA-gen duplicaten in het menselijk genoom. Ik zou verwachten dat de kern van het coderingssysteem
van het menselijke genoom goed 'beveiligd' was, maar de redundantie van gedupliceerde tRNA genen kent zijn grenzen. Die grenzen zijn bereikt in de Volendammer ziekte. Uiteindelijk is niets veilig voor mutatie. Er bestaat geen 100%
fault-tolerant, fail-safe ontwerp.
Achteraf gezien is het helemaal niet zo verwonderlijk dat
tRNA genen in meerdere kopieën aanwezig zijn in het genoom. Dat
geldt voor zoveel genen. Toch heb ik altijd gedacht dat je voor 61 codons hoogstens 61 tRNAs nodig zou hebben. Die 506 tRNA genen zijn voor mij een grote verrassing [4]. Achteraf is het ook niet zo verwonderlijk dat tRNA genen
introns bevatten, want de meerderheid van onze genen bevat introns. Dus waarom zouden tRNA genen een uitzondering zijn? De mens heeft 'slechts' 687 intronloze genen. (In totaal heeft de mens 20.000 genen). Waarom hadden niet alle tRNA genen intronloos kunnen zijn? Dan was deze ellende niet opgetreden. Tenslotte is het achteraf niet zo heel verwonderlijk dat de 'Volendammer' introns een eigen splicing mechanisme hebben, want er zijn verschillende soorten introns die op verschillende wijze verwijderd worden.
Conclusie
Menselijk gezien is de Volendammer ziekte een tragedie. Een ernstige ziekte waarbij het kind gehandicapt ter wereld komt en die ook nog erfelijk is. Een troost is dat zowel het PCH2 dragerschap van de ouders, als de aanwezigheid van de ziekte bij de foetus prenataal getest kan worden. Uit de documentaire [1] blijkt dat ouders die reeds een gehandicapt kind hebben
hier dankbaar gebruik van maken.
Genetisch gezien geeft deze ziekte een ongekende blik in de fundamenten van de menselijke genetica en de foutgevoeligheid van de genetische code in het algemeen. Ik ken geen andere puntmutatie met zo'n desastreus, catastrofaal effect. Een simpele puntmutatie in een enkel enzym gooit het hele systeem van genetische codering van een groot aantal eiwitten overhoop. De schade die de Volendammer mutatie aanricht is groot genoeg voor het ontstaan van een permanente onherstelbare handicap, maar niet groot genoeg om dodelijk te zijn voor het embryo c.q. foetus. Het is geen wonder dat de betreffende mutatie in TSEN54 een ernstig gehandicapt kind oplevert, maar het is vooralsnog een raadsel waarom deze mutatie niet in een zeer vroeg stadium van de zwangerschap fataal is voor het embryo.
17 mrt: kleine edits en verbeterde figuur toegevoegd (met dank aan Marleen).
Noten
- De Volendammer ziekte KRO-NRCV Kruispunt zondag 21 februari 2016: "Het katholieke dorp Volendam leidt onder genetische ziektes waardoor gehandicapte kinderen geboren worden die veelal vroeg sterven. Het AMC heeft genetische spreekuur waar aanstaande ouders zich kunnen laten testen. In Kruispunt vertellen ouders van gehandicapte kinderen hun verhaal. Zo verloor de familie Veerman twee zoontjes aan de ziekte en stond voor de keus of ze nog een derde kind wilden."
- Frank Baas et al (2011) Classification, diagnosis and potential mechanisms in Pontocerebellar Hypoplasia, Orphanet Journal of Rare Diseases 2011.
- Pontocerebellaire hypoplasie type 2 (Nederlandse wikipedia)
- Ik vraag me af: hoe houd je copieën van genen identiek aan elkaar? waarom gaan ze niet in de loop van de evolutie divergeren van elkaar? en dat laatste wil je toch niet? het gaat hier tenslotte om de genetische code. [23 maart]







